













What does the FDA’s final rule on LDTs change?
The rule makes it explicit that in vitro diagnostics (IVDs) that are manufactured by clinical laboratories—i.e., laboratory-developed tests (LDTs)—are considered devices under the Federal Food, Drug, and Cosmetic Act (FDCA). IVDs are tests performed on samples such as blood or tissue that have been taken from the human body. LDTs are designed and used within a single laboratory that is certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA).
In the final rule, rather than creating a definition of LDTs, the FDA added 10 words to the regulatory definition for IVD products.
In vitro diagnostic products are those reagents, instruments, and systems intended for use in the diagnosis of disease or other conditions, including a determination of the state of health, in order to cure, mitigate, treat, or prevent disease or its sequelae. Such products are intended for use in the collection, preparation, and examination of specimens taken from the human body. These products are devices as defined in section 201(h) of the Federal Food, Drug, and Cosmetic Act (the act), and may also be biological products subject to section 351 of the Public Health Service Act, including when the manufacturer of these products is a laboratory.
When will the changes take effect?
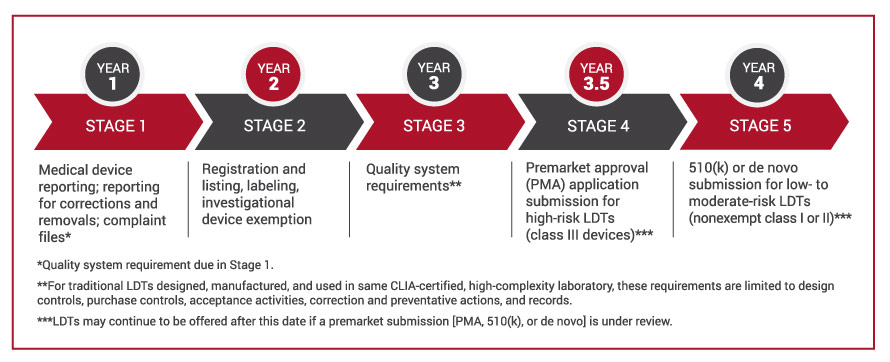
The rule was published in the Federal Register on May 6, 2024. The FDA established a four-year, five-stage “phaseout policy” during which medical device law is phased in for LDTs.
Key Dates:
Stage 1—May 6, 2025
Stage 2—May 6, 2026
Stage 3—May 6, 2027
Stage 4—November 6, 2027
Stage 5—May 6, 2028
What are the policies?
The FDA will phase out its general enforcement discretion approach for LDTs so that IVDs “manufactured” by a laboratory will generally fall under the same enforcement approach as other IVDs. The FDA is adopting targeted enforcement discretion policies for specific categories of IVDs manufactured by a laboratory.
Which categories of IVDs remain under the targeted enforcement discretion policy?
- “1976-type” LDTs, which have the following characteristics:
- Use manual techniques (without automation and without the use of software) that are performed by laboratory personnel with specialized expertise
- Use components legally marketed for clinical use
- Are designed, manufactured, and used within a single CLIA-certified laboratory that meets the requirements under CLIA for high-complexity testing
- Examples of 1976-type IVDs include certain immunohistochemistry tests, cystic fibrosis sweat tests, certain colorimetric newborn screening tests, karyotypic tests.
- Human leukocyte antigen (HLA) tests for transplant, which have the following characteristics:
- Are designed, manufactured, and used within a single CLIA-certified laboratory that meets the requirements under CLIA for high-complexity histocompatibility testing when used in connection with organ, stem cell, and tissue transplantation to perform HLA allele typing for HLA antibody screening and monitoring, or for conducting real and “virtual” HLA cross-match tests
- Forensic tests
- Tests intended solely for law enforcement
- Department of Defense (DoD) or Veterans Health Administration (VHA) tests
- Public health surveillance tests
Which other tests are subject to enforcement discretion?
- Currently marketed LDTs: LDTs that were marketed before May 6, 2024, that are not modified, or that are modified in certain limited ways
- New York State Clinical Laboratory Evaluation Program (NY CLEP)-approved LDTs: LDTs approved by the NY CLEP will generally not be subject to premarket review requirements.
- “Unmet needs” LDTs: LDTs manufactured and performed by a laboratory integrated within a healthcare system to meet an unmet need for patients within the same healthcare facility
- The FDA does not consider this to include patients who are being treated at an affiliated hospital with a different corporate ownership than the laboratory.
- “Unmet need” means that there is no available FDA-authorized IVD that meets the patient’s needs. Example scenarios include rare diseases, or when the FDA-authorized IVD is not indicated for use on the patient, or when the FDA-authorized IVD is not available to the patient.
What are the benefits of NY CLEP approval?
NY CLEP approval used to mean that labs could test patients in New York with the approved test. Under the FDA rule, New York approval still means that, but it also means labs can avoid submitting a test to the FDA for premarket notification or approval. It is a faster and much less expensive pathway. NY CLEP inspects laboratories on a regular basis, like the College of American Pathologists (CAP) does to ensure labs are meeting CLIA requirements, and reviews validation data for each test that the laboratory would like to offer in New York.
Were some tests grandfathered?
Not completely. For example, currently marketed LDTs are subject to listing and labeling requirements within two years, and monitoring going forward. Although currently marketed LDTs are not required to be submitted to the FDA for review and approval, meeting the listing and labeling requirements will still require a significant amount of work for many clinical laboratories.
What are the legal challenges to the FDA rule?
- The American Clinical Laboratory Association (ACLA) filed a lawsuit on May 29, 2024, in U.S. District Court for the Eastern District of Texas challenging the FDA’s authority to regulate LDTs as medical devices under the Federal Food, Drug, and Cosmetic Act (FDCA). (ACLA’s press release and supplemental material are linked here.) ACLA advocates for a collaborative approach to additional regulation as an alternative. ARUP filed a declaration supporting ACLA’s lawsuit, which is linked here.
- The Association for Molecular Pathology (AMP) filed a lawsuit on August 19, 2024, in U.S. District Court for the Southern District of Texas, which also challenges the FDA’s authority to regulate LDTs as medical devices and cited the potential impact of the rule on molecular diagnostics and precision medicine. AMP’s resource page, which includes a press release and the complaint, is linked here.
- On September 20, 2024, a judge consolidated the two cases in U.S. District Court for the Eastern District of Texas. A motion for summary judgment initiated by ACLA is pending, with final court filings in response to that motion due by December 31, 2024.
How does the Supreme Court decision on the so-called Chevron doctrine affect the lawsuits?
In June, the U.S. Supreme Court handed down a decision in the landmark Loper Bright Enterprises v. Raimondo case that is generally viewed as favorable to the ACLA and AMP challenges to the FDA’s rule because the decision overturned the so-called Chevron doctrine. That 40-year-old legal precedent instructed courts to defer to federal agencies’ authority in reasonable interpretations of ambiguous laws. However, even if ACLA and AMP prevail in federal court, any decision could be appealed.
What is ARUP doing to help labs?
ARUP maintains and updates a free resource library of webinars, podcast episodes, articles, and other materials to help clients and labs understand the FDA’s rule (linked here). Our mission has always been to share knowledge and information, and we will help clients with anything they need while continuing to advocate for labs.